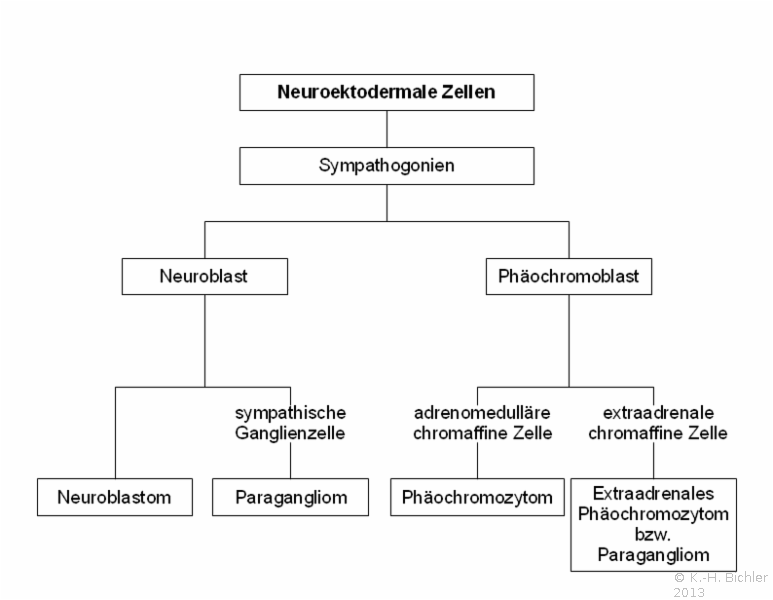
- Abbildung HG1:
- Embryogenese des Neuroblastoms
- (Mod. n.
 Literatur:Kaul, E.: http://edoc.ub.uni-muenchen.de/707/1/Kaul_Elisabeth.pdf, Oktober 2011)
Literatur:Kaul, E.: http://edoc.ub.uni-muenchen.de/707/1/Kaul_Elisabeth.pdf, Oktober 2011)
Das Neuroblastom ist der häufigste solide Tumor beim Säugling und im Kindesalter. Er macht 50% der bösartigen Tumoren bei Neugeborenen aus. Die Tumoren gehen von sympathischen Ganglien bzw. der Medulla der Nebenniere aus. Von letzterer stammen 2/3 der Tumoren. Fernerhin sind die sympathischen Ganglien der paravertebralen Leiste des hinteren Mediastinums sowie paravertebral im unteren Abdomen Ausgangspunkte des Tumors (;) Abbildung HG1). Als Tumoren des peripheren sympathischen Nervensystems können sie Katecholamine produzieren.
Abbildung HG1). Als Tumoren des peripheren sympathischen Nervensystems können sie Katecholamine produzieren.
Der Tumor weist im Säuglingsalter andere Eigenschaften als bei Kleinkindern auf. Dabei sind die Tumoren in ihrer Beschaffenheit in allen Altersstufen gleich. Bei Säuglingen kommt es in der Regel bei Generalisierung nicht zu Knochenläsionen, was bei Kindern älter als 1 Jahr, der Fall ist. Bei Säuglingen ist eine spontane Regression möglich, nicht jedoch bei älteren Kindern. Metastasen (lymphogen und hämatogen) finden sich in Leber und Haut (neugeborene Säuglinge) in Lunge und Knochen u.z. Schädel und Orbita bei älteren Kindern.
Man unterscheidet verschiedene klinisch-biologische Gruppen des Neuroblastoms. Es finden sich solche, die selten zum Tode führen (Säuglinge) und andere (>1Jahr), die einen raschen tödlichen Verlauf nehmen, trotz aller therapeutischen Fortschritte (Chemotherapie).
Diese biologischen Eigenschaften stehen im Zusammenhang mit ![]() AmplifikationenGenamplifikationSelektive Vervielfachung eines bestimmten Gens. Z.B. N-MYC-Onkogen: 3 zu 300 Kopien beim Neuroblastom. Hier gibt es eine strenge Korrellation zwischen N-MYC-Amplifikation, fortgeschrittenem Stadium und schlechter Prognose des N-MYC-
AmplifikationenGenamplifikationSelektive Vervielfachung eines bestimmten Gens. Z.B. N-MYC-Onkogen: 3 zu 300 Kopien beim Neuroblastom. Hier gibt es eine strenge Korrellation zwischen N-MYC-Amplifikation, fortgeschrittenem Stadium und schlechter Prognose des N-MYC-![]() OnkogensOnkogenentwicklungOnkogene entwickeln sich Protoonkogenen. Strukturdefekte oder Expression führen zu Störungen im Zellstoffwechsel, derartig veränderte Gene werden als Onkogene bezeichnet.
OnkogensOnkogenentwicklungOnkogene entwickeln sich Protoonkogenen. Strukturdefekte oder Expression führen zu Störungen im Zellstoffwechsel, derartig veränderte Gene werden als Onkogene bezeichnet.
![]()
Hentze, M.W. et al: "Einführung in die Medizinische Molekularbiologie", Springer Berlin, 1990
.
Bei den N-MYC-Onkogenen handelt es sich um Gene, die für die Zellproliferation und Gewebedifferenzierung von Bedeutung sind und bei denen Defekte in der Struktur oder Expression entstanden sind. Dadurch treten Störungen im Zellmetabolismus auf, die als Teilschritte der Tumorentwicklung zu verstehen sind ![]() Literatur:Hentze, M. W., Kuiozi, A. E., Bartram, C. R.: "Einführung in die Medizinische Molekularbiologie", Springer Berlin, 1990.
Literatur:Hentze, M. W., Kuiozi, A. E., Bartram, C. R.: "Einführung in die Medizinische Molekularbiologie", Springer Berlin, 1990.
![]() Drei genetisch verschiedene TypenDrei genetisch verschiedene Typen1. Tumoren ohne N-MYC-Amplifikation, hyperdiploide bzw. triploide - DNA.
Drei genetisch verschiedene TypenDrei genetisch verschiedene Typen1. Tumoren ohne N-MYC-Amplifikation, hyperdiploide bzw. triploide - DNA.
Patienten dieser Gruppe sind zumeist jünger als 1 Jahr und im Stadium I oder II oder IVs mit guter Prognose.
2. Tumoren, bei denen ein Verlust an Heterozygotie im Chromosom 1p zu 50% besteht. Die TRK-A-Expression ist schwach, eine N-MYC-Amplifikation fehlt, diploide bzw. tetraploide DNA.
Bei diesen Kindern > 1 Jahr und fortgeschrittenen Tumorstadien besteht in 25% bis 50% eine Dreijahresüberlebensrate.
3. Hier handelt es sich um Tumoren mit N-MYC-Amplifikation, fehlender TRK-A-Expression und Verlust der Heterozygotie (80-90%) in Chromosom 1 (TRK = Onkogen beim Schilddrüsenkarzinom). Diese Patienten sind zwischen 1 und 5 Jahren in fortgeschrittenen Krankheitsstadien, rascher Tumorexpression und schlechter Prognose. von Neuroblastomen sind auch im klinischen Verlauf zu unterscheiden ![]() Literatur:Humpl, T., Gutjahr, P.: "Paediatric oncology", Curr Opin Urol, 8, 525-528, 1998
Literatur:Humpl, T., Gutjahr, P.: "Paediatric oncology", Curr Opin Urol, 8, 525-528, 1998
Brodeur, G.M. et al: "International Criteria for Diagnosis, Staging, and Response to Treatment With Neuroblastoma", J Clinical Oncology, 6, 1874-1881, 1988
Bartram, C. R. et al: "Klinische Molekularbiologie" in Hentze, M. W., Kulozi, A. E.: "Einführung in die Medizinische Molekularbiologie", Springer Berlin, 1990
.